Fighting Painful Misconceptions About Sickle Cell Disease In The ER
When sickle cell patients arrive at emergency rooms, they often have great difficulty getting the treatment they need. Paula Tanabe, an associate professor at the Duke University School of Nursing, is making it her mission to change that.
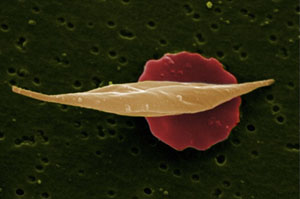
Photo by Wellcome Images via Flickr
Sickle cell disease, an incurable genetic blood disorder most common among people of African descent, affects 100,000 Americans. It causes normally disc-shaped red blood cells to take the form of pointed crescents or sickles. Common complications — which often bring death by the mid-40s — include strokes, anemia, severe infections and damage to blood vessels, kidneys, eyes, and other organs. Treatment can also be complicated because many patients are unable to work and that can lead to a lack of health insurance.
Acute and unpredictable pain, severe enough to require large doses of narcotics, is a well-documented feature of sickle cell disease. But Tanabe has found that too often patients in sickle-cell agony are not handled properly by doctors and nurses in the emergency department because they erroneously think the patients may be running scams, seeking drugs.
Patients suffering recurrent episodes know what works for their pain, but health care personnel widely believe that knowledge is a sign of addiction. Patients with sickle cell pain wait an average of 30 minutes longer for pain medication than people with other extremely painful conditions, such as kidney stones, Tanabe’s research shows.

Paula Tanabe
She believes stereotyping is the number one barrier behind that wait: “After being cute little kids with sickle cell that everybody felt bad for when they had pain, now they become this African American 20-year-old asking for high doses of opioid narcotics.”
Tanabe says better training of health providers and changes in emergency room procedures can improve the care that sickle cell patients receive. She spoke recently with KHN, and here is an edited transcript of the interview.
Q. What do patients typically experience during pain crises?
A. What brings people to the emergency room is a crisis in which the blood can’t deliver oxygen to the extremities. It is a sudden onset of severe, excruciating pain, which people describe as feeling as if all their bones are breaking. Others describe it as an hourglass and their blood is “stuck.” The pain is so bad and sudden that people require high doses of opioids. Patients who have a regular health care provider and have chronic pain are usually prescribed oral opioids to have at home. They take pain pills by mouth. [But] at the time of a really bad crisis, the pills don’t work any more. People need to come in for intravenous pain medication. They usually try to avoid coming into the ER at all costs because they just have to wait a long time.
Q. Are people with sickle cell likely to be addicted?
A. There are no data to support that sickle cell patients have any more likelihood of being addicted than anybody else.
Q. You compared the treatment in emergency rooms of patients with kidney stone pain and with sickle cell disease. What did you find?
A. A lot of white people get kidney stones and that pain is really, really horrible. Even though sickle cell patients had higher pain scores and were triaged appropriately higher, they still waited longer.
Racism and the disease stigma itself are two barriers that you just can’t get away from. Clearly we can’t pretend that racism doesn’t play some part in this. If this were a white disease, people still wouldn’t be dying in their forties. That’s the bottom line. Sickle cell was discovered 102 years ago and there is only one drug, hydroxyurea, and blood transfusions to treat it.
Q. So you believe racism has discouraged progress in treating sickle cell?
A. It is a rare disease. However, there are other rare genetic diseases that get a lot more attention and people are not afraid to talk about. Cystic fibrosis is a good example, and way more rare than sickle cell. It’s a white disease. There is some attention to sickle cell, but it needs more dollars and more research. There are very few hematologists, very few providers across the country, who specialize in it. Patients do need a good hematologist or sickle cell expert to take care of their disease.
There has been progress. People didn’t used to live to be adults. Kids would die of stroke or of really bad infections before they were 20, and in some countries they still do. The fact that we have adults is a good thing. All kids are screened at birth in the United States. Once you identify a child, they go on penicillin until they’re five, and then they don’t get the really bad infections and die from that.
Q. You have done research that documents the problem. Your current research involves methods of improving treatment. What does that involve?
A. We are implementing some education and some analgesic protocols. We hope this will help lower the time needed for patients to get analgesia, or pain control, and admission rates. We’ve administered a baseline attitude survey to providers. We will administer it again. We’d like to see improvement in clinician attitude. We’re really looking at patient satisfaction, provider attitudes, and then a lot of efficiency outcomes. Another aim of our grant is to put a toolbox together of all the protocols and the educational materials that we’ve developed and make it available.