There’s no doubt that surgically implanted medical devices can improve lives.
Hip and knee replacements can help people regain their mobility. Drug pumps can deliver doses of pain-relieving medicine on demand. And metal rods can stabilize spines and broken bones.
But implanted devices can also do serious damage, as happened to Mechel Keel, who lives in Owosso, Mich.
To fix her leaky bladder, an OB-GYN stitched a flexible mesh strap inside her pelvis in 2004. But within months, the mesh hardened and started cutting her insides.
The pain kept her from returning to her job as a hairdresser. The injuries and scar tissue that developed required multiple surgeries to correct and also resulted in chronic infections.
Keel said she understands why her doctor in Tennessee thought the high-tech mesh would help. But she also now feels she was treated like “a guinea pig.”
“We were the testers,” she said. “There was no animal testing done. We were the animals.”
Thousands of cases of complications from surgical mesh have been reported to the Food and Drug Administration. More and more ailments are being treated these days with medical devices, including implants. And most of these medical devices, unlike pills in a medicine cabinet, don’t go through human testing before being offered to patients.
But some devices break down or malfunction in people’s bodies, and reports about sometimes debilitating injuries have led the FDA to rethink how it assesses medical devices before allowing them to be sold.
For devices in which failure is obviously life-threatening, regulators have required some sort of human testing as part of the most stringent path — known as premarket approval. But most medical devices enter the market after manufacturers provide technical information and show that the devices are similar to others that have been legally sold previously.
The FDA has acknowledged that some seemingly safe devices have caused major problems, and the agency has elevated the risk level of those products following reports of injuries, as it did with urogynecologic surgical mesh for some uses in 2016. “Unfortunately, the FDA cannot always know the full extent of the benefits and risks of a device before it reaches the market,” the agency said in a recent statement.
“We have things like metal-on-metal hips,” said health journalist Jeanne Lenzer about a kind of orthopedic implant. “Outside the body, [they] seem to function just fine. They put them in little machines, rock them back and forth — they don’t break. [But] put them inside people, and something very different happens.”
There have been massive recalls of hip implants, for example, due to devices causing swelling and pain. And there have been problems with weakened bones in patients who received hip implants that contained plastic.
Lenzer wrote a scathing book, “The Danger Within Us,” about the device industry and said she was “dumbfounded” to find out how many devices never went through human testing the way drugs do.
That’s in part because of a regulatory review process known as 510(k) for a section of the FDA law covering medical devices. Manufacturers typically show their product has “substantial equivalence” to a “predicate device” that has already been legally marketed.
That standard can perpetuate problems. “You just say your device is like an old device, and the old device was never tested, nor was your device,” Lenzer said.
In practice, sometimes the basis for a whole family tree of devices turns out to be defective. Pelvic mesh is a relevant example, with much of what’s on the market being based on mesh that was around prior to implementation of FDA regulations for medical devices in 1976. One study found that 16 percent of mesh on the market was designed like products that had been pulled from the market because of safety concerns.
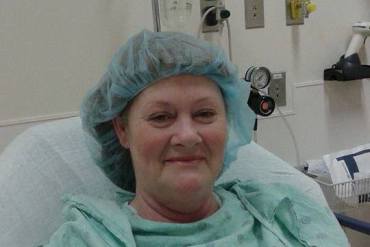
After she experienced bladder problems, Mechel Keel of Owosso, Mich., had flexible mesh implanted. But the mesh hardened inside her, causing pain and infections. In this 2015 photo, she is awaiting surgery to remove parts of the failed mesh.(Courtesy of Mechel Keel)
Thousands of women have filed suit — or reached settlements — with medical device companies that manufacture pelvic mesh.
“I would want nothing if you could just give me my life back,” said Gloria Jones of Hillsdale, Mich., who is one of thousands who have settled with device manufacturers over faulty mesh. “They could have given me millions, but all I needed was my life back.”
Jones, who has struggled to continue working through crippling abdominal pain as a middle school special-education teaching assistant, has had four surgeries to remove pieces and continues to require intravenous drugs to control infections.
“It seems like I get off one antibiotic, and three days later I have another one,” she said. “I would beg anybody who is even thinking of putting mesh in their bodies to stop and get a second opinion.”
In response to problems reported with mesh, the FDA started requiring human testing for some of these products in recent years. The agency held an advisory committee meeting on Feb. 12 to discuss the safety and effectiveness of mesh and how it should be regulated.
Manufacturers by and large have said they don’t oppose what the FDA is trying to do, calling the changes reasonable. But they have pushed back against calls to bring regulation of medical devices in line with that of pharmaceuticals.
“If you’re treating someone for high cholesterol, the testing that you go through to ensure safety and effectiveness on a chemical that’s going to be used in your body to control your cholesterol is just very different than it would be for the implantation of a heart valve,” said Scott Whitaker, CEO of AdvaMed, a trade association for medical device companies. “Honestly, it’s apples and oranges.”
Whitaker dismisses the idea that devices, which range from tongue depressors to surgical robots, should all go through human trials.
“Testing should be as complete and as thorough and as ethical and as appropriate as possible. But it doesn’t all fit the same and can’t all fit the same standard,” Whitaker said. “And while we always strive for 100 percent, there are times when something might not go according to plan. It could also be because the surgery didn’t go as was planned.”
The FDA declined NPR’s request for an interview. But the agency has released some written justification for the regulatory revamp.
“We believe firmly in the merits of the 510(k) process,” FDA Commissioner Scott Gottlieb said in a November statement, noting that applications have more than doubled in size to an average of 1,185 pages. “But we also believe that framework needs to be modernized to reflect advances in technology, safety and the capabilities of a new generation of medical devices.”
In the same statement, the agency addressed some specific shortcomings and charted plans to make changes to the process over the next few months:
- Pushing back on manufacturers that base any new device’s marketing application on one that’s more than 10 years old;
- More actively watching how devices perform once they’re on the market, rather than relying on patients to report problems;
- Scrapping the 510(k) name for something more descriptive, such as the “Safety and Performance Based Pathway.”
Dr. Michael Matheny, a Vanderbilt University professor who tracks medical devices, approves of the FDA’s incremental approach and calls it thoughtful.
“It would really be unfortunate if patients wouldn’t consider any medical devices at all to be used in their bodies,” Matheny said. “But I do think being aware that there’s nothing without risk is also important.”
Matheny notes, though, that in some ways the risks can be more profound for devices than medication. If the FDA recalls pills, a patient can at least stop taking them immediately, he said. With implanted devices, patients are sort of stuck, at least for a while — and that’s if surgeons can even safely remove them.
This story is part of a partnership that includes Nashville Public Radio, NPR and Kaiser Health News.



