
NeDina Brocks-Capla sits in her kitchen in San Francisco. Her son Kareem Jones died at age 36 from sickle cell disease. (Jenny Gold/KHN)
For more than a year, NeDina Brocks-Capla avoided one room in her large, brightly colored San Francisco house — the bathroom on the second floor.
“It was really hard to bathe in here, and I found myself not wanting to touch the walls,” she explained. The bathroom is where Brocks-Capla’s son Kareem Jones died in 2013 at age 36, from sickle cell disease.
It’s not just the loss of her son that upsets Brocks-Capla; she believes that if Jones had gotten the proper medical care, he might still be alive today.
Sickle cell disease is an inherited disorder that causes some red blood cells to bend into a crescent shape. The misshapen, inflexible cells clog the blood vessels, preventing blood from circulating oxygen properly, which can cause chronic pain, multi-organ failure and stroke.
About 100,000 people in the United States have sickle cell disease, and most of them are African-American.
Patients and experts alike say it’s no surprise then that while life expectancy for almost every major malady is improving, patients with sickle cell disease can expect to die younger than they did 20 years ago. In 1994, life expectancy for sickle cell patients was 42 for men and 48 for women. By 2005, life expectancy had dipped to 38 for men and 42 for women.
Sickle cell disease is “a microcosm of how issues of race, ethnicity and identity come into conflict with issues of health care,” said Keith Wailoo, a professor at Princeton University who writes about the history of the disease.
It is also an example of the broader discrimination experienced by African-Americans in the medical system. Nearly a third report that they have experienced discrimination when going to the doctor, according to a poll by NPR, Robert Wood Johnson Foundation and Harvard T.H. Chan School of Public Health.
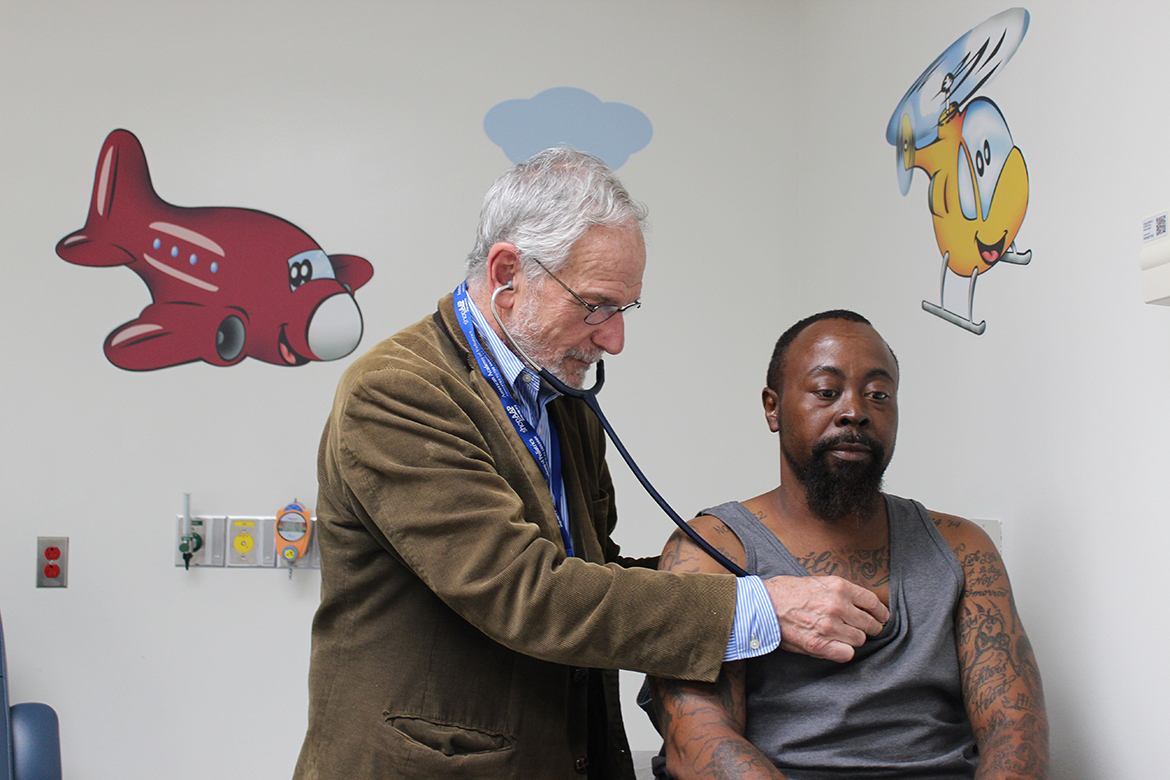
Dr. Elliott Vichinsky examines Derek Perkins at the sickle cell center at UCSF Benioff Children’s Hospital Oakland, which sees both children and adults. (Jenny Gold/KHN)
“One of the national crises in health care is the care for adult sickle cell,” said leading researcher and physician Dr. Elliott Vichinsky, who started the sickle cell center at UCSF Benioff Children’s Hospital Oakland in 1978. “This group of people can live much longer with the management we have, and they’re dying because we don’t have access to care.”
Indeed, with the proper care, Vichinsky’s center and the handful of other specialty clinics like it across the country have been able to increase life expectancy for sickle cell patients well into their 60s.
Vichinsky’s patient Derek Perkins, 45, knows he has already beaten the odds. He sits in an exam room decorated with cartoon characters at Children’s Hospital Oakland, but this is the adult sickle cell clinic. He’s been Vichinsky’s patient since childhood.
“Without the sickle cell clinic here in Oakland, I don’t know what I would do. I don’t know anywhere else I could go,” Perkins said.
When Perkins was 27, he once ended up at a different hospital where doctors misdiagnosed his crisis. He went into a coma and was near death before his mother insisted he be transferred.
“Dr. Vichinsky was able to get me here to Children’s Hospital, and he found out what was wrong and within 18 hours — all I needed was an emergency blood transfusion and I was awake,” Perkins recalls.
Kareem Jones lived just across the bay from Perkins, but he had a profoundly different experience.
Jones’ mother, Brocks-Capla, said her son received excellent medical care as a child, but once he turned 18 and aged out of his pediatric program, it felt like falling off a cliff. Jones was sent to a clinic at San Francisco General Hospital, but it was open only for a half-day, one day each week. If he was sick any other day, he had two options: leave a voicemail for a clinic nurse or go to the emergency room. “That’s not comprehensive care — that’s not consistent care for a disease of this type,” said Brocks-Capla.
Brocks-Capla is a retired supervisor at a worker’s compensation firm. She knew how to navigate the health care system, but she couldn’t get her son the care he needed. Like most sickle cell patients, Jones had frequent pain crises. Usually he ended up in the emergency room where, Brocks-Capla said, the doctors didn’t seem to know much about sickle cell disease.
When she tried to explain her son’s pain to the doctors and nurses, she recalled, “they say have a seat. ‘He can’t have a seat! Can’t you see him?’”
Studies have found that sickle cell patients have to wait up to 50 percent longer for help in the emergency department than other pain patients. The opioid crisis has made things even worse, Vichinsky added, as patients in terrible pain are likely to be seen as drug seekers with addiction problems rather than patients in need.
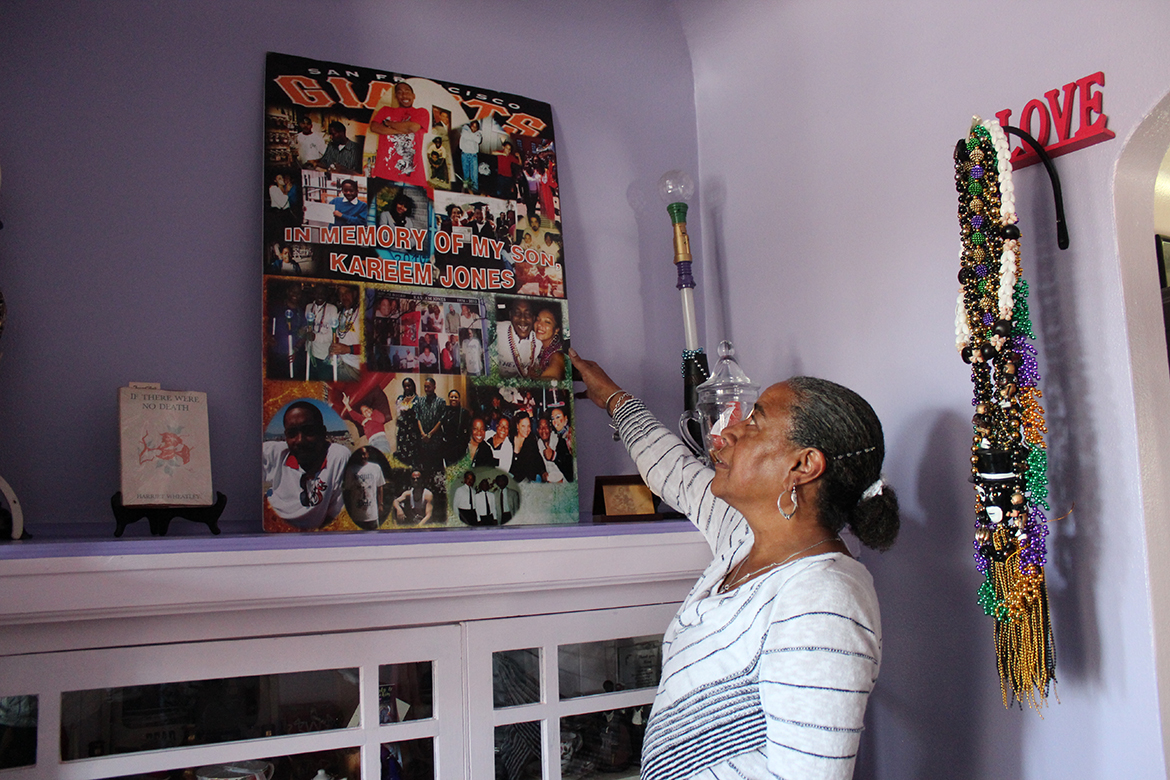
NeDina Brocks-Capla stands in her living room in San Francisco. She made a shrine filled with memories of son Kareem Jones, who died of sickle cell disease in 2013. (Jenny Gold/KHN)
Despite his illness, Jones fought to have a normal life. He lived with his girlfriend, had a daughter and worked as much as he could between pain crises. He was an avid San Francisco Giants fan.
For years, he took a drug called hydroxyurea, but it had side effects, and after a while Jones had to stop taking it. “And that was it, because you know there isn’t any other medication out there,” said Brocks-Capla.
Indeed, hydroxyurea, which the FDA first approved in 1967 as a cancer drug, was the only drug on the market to treat sickle cell during Jones’ lifetime. In July, the FDA approved a second drug, Endari, specifically to treat patients with sickle cell disease.
Funding by the federal government and private foundations for the disease pales in comparison to other disorders. Cystic fibrosis offers a good comparison. It is another inherited disorder that requires complex care and most often occurs in Caucasians. Cystic fibrosis gets seven to 11 times more funding per patient than sickle cell disease, according to a 2013 study in the journal Blood. From 2010 to 2013 alone, the FDA approved five new drugs for the treatment of cystic fibrosis.
“There’s no question in my mind that class and color are major factors in impairing their survival. Without question,” Vichinsky said of sickle cell patients. “The death rate is increasing. The quality of care is going down.”
Without a new medication, Jones got progressively worse. At 36, his kidneys began to fail, and he had to go on dialysis. He ended up in the hospital, with the worst pain of his life. The doctors stabilized him and gave him pain meds but did not diagnose the underlying cause of the crisis. He was released to his mother’s care, still in incredible pain.
At home, Brocks-Capla ran him a warm bath to try to soothe his pain and went downstairs to get him a change of clothes. As she came back up the stairs, she heard loud banging against the bathroom walls.
“So I run into the bathroom and he’s having a seizure. And I didn’t know what to do. I was like, ‘Oh come on, come on. Don’t do this. Don’t do this to me.’”
She called 911. The paramedics came but couldn’t revive him. “He died here with me,” she said.
It turned out Jones had a series of small strokes. His organs were in failure, something Brocks-Capla said the hospital missed. She believes his death could have been prevented with consistent care — the kind he got as a child. Vichinsky thinks she is probably right.
“I would say 40 percent or more of the deaths I’ve had recently have been preventable — I mean totally preventable,” he said, but he got to the cases too late. “It makes me so angry. I’ve spent my life trying to help these people, and the harder part is you can change this — this isn’t a knowledge issue. It’s an access issue.”
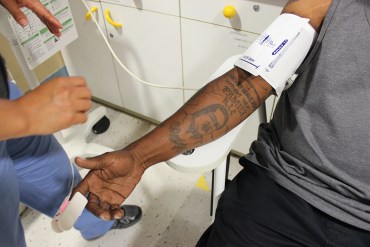
A nurse takes patient Derek Perkins’ blood pressure at the sickle cell center at UCSF Benioff Children’s Hospital Oakland — one of only three places in California that offer specialized services for adults with sickle cell disease. (Jenny Gold/KHN)
Vichinsky’s center and others like it have made major advances in screening patients for the early signs of organ failure and intervening to prevent premature death. Patients at these clinics live two decades longer than the average sickle cell patient.
Good care for sickle cell requires time and training for physicians, but it often doesn’t pay well, because many patients are on Medicaid or other government insurance programs. The result is that most adult sickle cell patients still struggle even to access treatments that have been around for decades, Vichinsky said.
The phenomenon is nothing new — the disease that used to be known as sickle cell anemia has had a long and sordid past. It was first identified in 1910 and helped launch the field of molecular biology. But most of the research was used to study science rather than improving care for sickle cell patients, Vichinsky said.
In the 1960s and ’70s, sickle cell became a lightning rod for the civil rights movement. At the time, the average patient died before age 20. The Black Panther Party took up the cause and began testing people at their “survival conferences” across the country.
“I’m sure we tested over four-and-a-half-thousand people for sickle cell anemia last night — and I think that the voter registration is running neck and neck with it,” Black Panther Party Chairman Bobby Seale told news crews at an event in Oakland in 1972.
The movement grew, and Washington listened. “It is a sad and shameful fact that the causes of this disease have been largely neglected throughout our history,” President Richard Nixon told Congress in 1971. “We cannot rewrite this record of neglect, but we can reverse it. To this end, this administration is increasing its budget for research and treatment of sickle cell disease.”
For a while, funding did increase, newborn screening took hold and by the 1990s, life expectancy had doubled, with patients living into their 40s. But over time, funding waned, clinics closed, and life expectancy started dropping again.
Vichinsky pushes against that trend for patients like Derek Perkins. The father of four looks healthy and robust, but like most sickle cell patients, he has episodes of extreme pain and has problems with his kidneys, heart, hips and breathing. Keeping him thriving requires regular checkups and constant monitoring for potential problems.
“The program Dr. Vichinsky is running here, I feel I owe my life to [it],” said Perkins. “If it wasn’t for him and the things that he did for me, my family wouldn’t have me.”
KHN’s coverage of children’s health care issues is supported in part by a grant from The Heising-Simons Foundation.



