In some medical device recalls, including some of the most serious, the FDA and the manufacturers let doctors and hospitals continue to use the devices. (Sarah Silbiger/Getty Images)
In 2016, medical device giant Abbott issued a recall for its MitraClip cardiac device — “a Class I recall, the most serious type,” the FDA said.
“Use of this device may cause serious injuries or death,” an FDA notice about the recall said.
But neither the manufacturer nor the FDA actually recalled the device or suspended its use. They allowed doctors to continue implanting the clips in leaky heart valves in what has become a common procedure.
In a notice, the manufacturer explained, “Abbott is not removing product from commercial distribution.” Rather, Abbott revised instructions for use and required doctors who implant the clips to undergo training.
When it comes to medical devices, recalls can include not only “removals,” in which the device is removed from where it is used or sold, but also “corrections,” which address the problem in the field — for instance, by repairing, adjusting, relabeling, or inspecting a device.
“It’s very oxymoronic,” said Rita Redberg, a cardiologist at the University of California-San Francisco and former editor-in-chief of the journal JAMA Internal Medicine. “A recall makes it sound like it’s recalled. But that is not actually what it means.”
Though the FDA and federal regulations call these actions recalls, they might be described more aptly as “non-recalls.” And they have happened repeatedly in recent years. For instance, in addition to other Abbott devices, products made by Medtronic, Abiomed, and Getinge have had recalls that left them in use.
Email Sign-Up
Subscribe to KFF Health News' free Morning Briefing.
Safeguarding the Public
Recalls that leave what the FDA identifies as potentially dangerous products in the marketplace can raise the question: Do they do enough to protect the public?
There are other ways to handle recalls. In announcements about products as varied as crib bumpers, pool drain covers, bicycle helmets, and coffee mugs, the Consumer Product Safety Commission routinely alerts consumers to stop using recalled products and contact the manufacturers for refunds, repairs, or replacements. The National Highway Traffic Safety Administration regularly advises consumers to bring recalled cars back to the dealer to have them fixed. When the U.S. Department of Agriculture and the FDA announce food recalls, they routinely tell consumers to return or discard the food.
In some cases, a medical device that is the subject of a recall can be kept on the market safely because there is a simple fix, said Sanket Dhruva, a cardiologist and an associate professor at UCSF who has studied FDA oversight of devices. In other cases, recalls that don’t remove devices from the market can provide unwarranted reassurance and leave the public at risk, Dhruva said.
From 2019 through 2023, there were 338 Class I medical device recalls, 164 of which were corrections and 174 of which were removals, FDA spokesperson Amanda Hils said.
Some products undergo recall after recall while they remain on the market. Products in the MitraClip line have been the subject of three rounds of recalls, none of which removed devices from use.
“When deciding whether a recall warrants device removal from the field, the FDA considers the frequency and severity of adverse events, effectiveness of the corrective actions that have been executed, and the benefits and risks of preserving patient access to the device,” FDA spokesperson Audra Harrison said.
The story of MitraClip, a device Dr. Oz helped invent to treat faulty heart valves, is a cautionary tale about the science, business, and regulation of medical technology.
Where recalled devices have already been implanted, “removal” doesn’t necessarily mean removing them from patients’ bodies. “When an implanted device has the potential to fail unexpectedly, companies often tell doctors to contact their patients to discuss the risk of removing the device compared to the risk of leaving it in place,” the FDA website says.
The FDA allowed the recalled MitraClip devices to remain in use “because the agency believed that the overall benefits of the device continued to outweigh the risks and the firm’s recall strategy was appropriate and adequate,” Harrison said.
The FDA reviews the recall strategies that manufacturers propose and often provides input to ensure the public will be protected, Hils said. The agency also monitors the effectiveness of recalls and, before terminating them, makes sure the strategy was carried out, Hils said.
Abbott, the maker of MitraClip, said the device has been proven safe and effective “based on more than 20 years of clinical evidence and has profoundly improved the lives of people living with mitral regurgitation,” a condition in which blood flows backward through the heart’s mitral valve. The condition can lead to heart failure and death.
“With MitraClip, we’re addressing the needs of people with MR who often have no other options,” company spokesperson Brent Tippen said.
Speaking of the MitraClip recalls, Redberg said, “So hard to imagine these are effective actions in protecting patients.”
In 2021, for Medtronic’s StealthStation S7 cranial software, the company and the FDA sent a different message.
StealthStation is an elaborate system of screens and other equipment that guides neurosurgeons using instruments in the brain — for instance, to biopsy or cut out tumors. Drawing from CT scans, MRIs, and other imaging, it’s meant to show the location of the surgical instruments.
In connection with a Class I November 2021 recall, the FDA website said potential inaccuracies in a biopsy depth gauge could result in “life-threatening injury (such as hemorrhage, unintended tissue damage, or permanent neurological injury), which could lead to death.”
The FDA website explained what Medtronic was doing about it.
“The recalling firm will provide a warning and instructional placard to be applied to impacted systems,” the website said. “Until a software update is available, ensure you are following the instructions below to prevent the issue from occurring,” it advised doctors.
In a statement to KFF Health News, Medtronic spokesperson Erika Winkels said the safety and well-being of patients is the company’s primary concern, and certain issues “can be safely and effectively remedied with a correction on site.”
Richard Everson, a neurosurgeon and an assistant professor at UCLA, noted that the 2021 recall allowed doctors to continue using unaffected StealthStation features, a benefit for patients and facilities depending on them.
“But, I mean, then you could ask, ‘Well, why don’t they just disable the view [of the brain] that’s bugged?’” Everson said. “Why would they give you the option of looking at an inaccurate one?”
“That’s kind of a strange solution,” he said.
The FDA lists the 2021 recall as still open, explaining “not all products have been corrected or removed.”
That recall was not the last word on problems with StealthStation. Since then, the manufacturer has submitted adverse event reports to the FDA describing trouble in cases involving various versions of StealthStation.
In a September 2022 case, guidance provided by a StealthStation device was allegedly off the mark, a procedure was aborted, and, when the patient awoke, they “had almost no speech for two days,” according to a Medtronic report. In the report, Medtronic said there was “insufficient information to determine the relationship of the software to the reported issue.”
In a February 2024 case, after brain surgery, an MRI found that the operation “missed the tumor” and that other tissue was removed instead, according to a report Medtronic submitted to the FDA. In the report, Medtronic said that when a company representative tested the system, it performed as intended.
In March 2024, Medtronic recalled versions of StealthStation S8 without removing them from hospitals. The company said at the time that it would provide a software update.
“Software updates are available to correct the anomalies identified in the 2021 S7 and 2024 S8 recalls and are actively being deployed,” Medtronic’s Winkels told KFF Health News in a July email. “While the software updates for the 2021 S7 recall are complete in the US, they remain ongoing in some international regions.”
In June 2023, Abiomed issued an urgent medical device correction for its Impella 2.5 intravascular micro axial blood pump, which supports the heart. In patients with a certain type of replacement heart valve, there was a risk of “destruction of the impeller blades,” which could cause “low flow” and “embolization of the fractured impeller material,” an entry on the FDA website said.
“Clinicians are cautioned to position the Impella system carefully in patients,” the FDA website said, among other instructions.
The updated instructions “provide technical guidance to mitigate the risk of rare complications,” Abiomed spokesperson Ryan Carbain said. There were no product removals and no reports of adverse events “related to product design or manufacturing,” Carbain said.
Another set of medical devices, Cardiosave Hybrid and Rescue Intra-Aortic Balloon Pumps made by Getinge of Sweden, have failed persistently, according to FDA records.
The devices — which are placed in the aorta, a major artery, to assist the heart — were the subject of eight Class I recalls from December 2022 to July 2023. All were corrections rather than removals, a KFF Health News analysis found.
In a May 2024 letter to health care providers, the FDA said that, in the previous 12 months, it had received almost 3,000 adverse event reports related to the balloon pumps. It was referring to reports of malfunctions and cases in which the products might have caused or contributed to a death or injury. Of those, 15 reportedly involved serious injury or death, the FDA said.
During the summer of 2023, the FDA noted that “alternative treatments are limited” and said the devices could continue to be used.
But, in May, the FDA changed its stance. The agency advised health care facilities to “transition away from these devices and seek alternatives, if possible.”
“These recommendations are based on our continued concerns” that the manufacturer “has not sufficiently addressed the problems and risks with these recalled devices.”
Getinge sent KFF Health News written answers from Elin Frostehav, the company’s president of Acute Care Therapies.
“There is no question that we would have liked to have solved these issues in full much earlier,” she said.
As a result of the FDA’s May action, the company “immediately paused proactive marketing” of the balloon pumps in the United States, and it is selling them only to customers who have no alternatives, Frostehav said.
“We are working with the agency to finalize remediation and product update solutions,” Frostehav said.
‘Known Possible Complications’
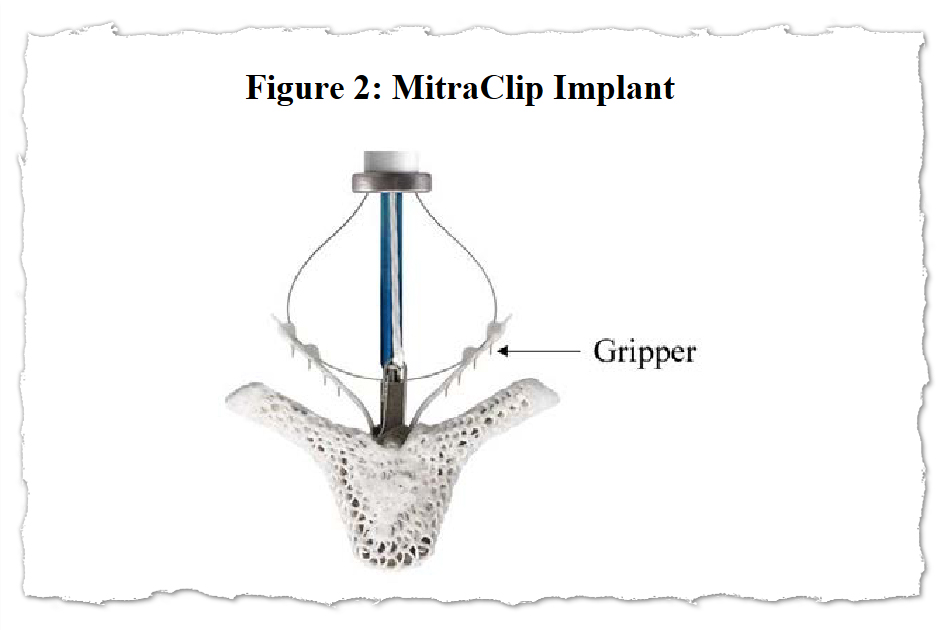
Abbott’s MitraClip system includes tiny clips implanted in the heart’s mitral valve and the equipment used to implant them. The apparatus features a steering mechanism with hand controls and a catheter that is threaded through a major vein, typically from an incision in the groin, to place one or more clips in the heart.
An image from the 2019 FDA document showing a clip implanted to hold flaps of the heart’s mitral valve together. MitraClip is deployed via a catheter threaded through a major blood vessel. (Photo illustration of 2019 FDA document)
Worldwide, more than 200,000 people have been treated with MitraClip, according to an Abbott website.
The 2016 MitraClip recall described cases in which “the user was unable to separate the implantable Clip from the delivery system.”
In a news release at the time, Abbott said it had “received a small number of reports” in which that happened.
Those cases “resulted in surgical interventions to remove the delivery system or replace the mitral valve, and it is expected that any future similar incidents would also require surgery to correct the problem,” the FDA said in a 2016 notice. “There was one patient death in these cases as a result of severe comorbidities following surgery.”
Years later, something similar happened.
In February 2021, a clip was implanted in an 81-year-old patient but the doctor couldn’t separate the clip from the delivery system, according to a report Abbott filed with the FDA. The patient was transferred to surgery, where the delivery system “had to be cut down in order to detach the clip.”
The patient then underwent an operation to replace the mitral valve, and, hours later, the patient was brought back to surgery to address bleeding, the report said.
The patient “coded” the next day and died from an aortic bleed, the report said.
In the report to the FDA, the manufacturer blamed “case-specific circumstances.”
“Cardiac arrest, hemorrhage and death are listed” in the device instructions “as known possible complications associated with mitraclip procedures,” the company said. “There is no indication of a product issue with respect to manufacture, design or labeling.”
The third MitraClip recall, initiated in September 2022, cited an “increase in clip locking malfunctions.”
Most of the reported malfunctions were not associated with adverse outcomes, the FDA said then. Treatment with MitraClip “remains within the anticipated risk levels,” the company told customers.
As with the two earlier recalls, the third advised doctors to follow the device’s instructions. But the 2022 recall identified a contributing factor: the way the device was made.
“Abbott has identified a contributing cause … as a change in the material properties of one of the Clip locking components,” the company said in a 2022 letter to customers.
“Abbott is working on producing new lots with updated manufacturing processing and raw material,” the company wrote. In the same letter, Abbott told doctors that, in the meantime, they could use the devices they had in stock.
Six days later, a clip opened while locked and a patient died, according to a report the manufacturer submitted to the FDA.
“There is no evidence that death was related to the device but it was likely related to the procedure,” Abbott wrote.
Now, almost two years later, the 2022 recall remains open, according to the FDA website, and “not all products have been corrected or removed.”
KFF Health News data editor Holly K. Hacker contributed to this report.
If you’ve had an experience with a medical device and would like to tell KFF Health News about it, click here to contact our reporting team.